近日,西安交大前沿院郭武生教授课题组在手性非环状α-胺基酮的合成方面取得重要进展。相关研究结果以“Propargylic Amination Enabled the Access to Enantioenriched Acyclic α-Quaternary α-Amino Ketones”为题发表在化学领域国际权威期刊 Journal of the American Chemical Society 。
手性α-胺基酮具有高反应活性的羰基和胺基,经过较为简单的后续化学反应,就可将其转化为多种复杂的手性胺类药物及中间体或生物活性分子。因此,手性α-胺基酮的高效多样化构筑是合成化学家们研究的重要目标之一。目前研究最多的构筑手性胺基酮的策略是利用酮的α-碳亲核进攻“氮正”试剂(一般为偶氮化物),再经过额外的一步或多步反应,将对应的产物转化为胺基酮。该方法主要的局限性有四个方面:一是该反应产物后续的转化步骤需要大量的还原试剂(一般为金属锌粉),从而导致一些安全隐患并伴随产生大量的固体废弃物;二是在肼基被还原为胺基的过程中,羰基也有可能被还原为醇,从而导致胺基酮的主体结构不能保持;三是该类方法的手性诱导步骤严重依赖于环状酮的优势构象,因此主要适用于环状手性α-胺基酮的合成;四是所合成的手性α-胺基酮产物上胺基的取代基种类较少,一般仅限于Boc或酯基。
商业化的自由胺种类繁多、取代基多样,是理想的构筑手性α-胺基酮的原料。然而以自由胺为原料通过形成C-N键的方式构筑手性α-胺基酮极具挑战性,主要原因是:1、潜在的胺酮缩合副反应会影响反应效率和底物普适性;2、酮的α-碳和胺都具有亲核性,因电性不匹配而导致其直接偶合变的异常困难;3、脂肪酮底物具有两个反应活性位点(α-碳和α'-碳),要同时控制胺化的区域选择性并保持高的手性选择性极其困难。
针对上述挑战,郭武生教授课题组设计了一种新型的环状碳酸酯。在铜的催化作用下,该碳酸酯经历脱羧反应生成与铜键合的两性烯醇离子中间体。该中间体含有α和β两个具有平面特征的sp2杂化碳,从而较大程度地减少了形成四取代立体中心的空间位阻效应。此外,烯醇氧负离子一是会与胺亲核试剂形成牢固的氢键作用,二是可作为潜在的π-π作用受体,这些因素都有利于胺试剂亲核进攻的不对称选择性的控制(图1)。
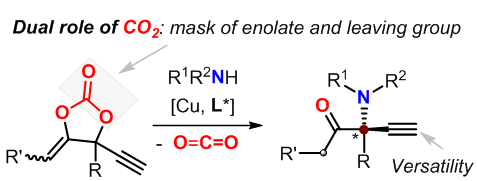
图1 铜催化的炔丙基胺化策略构筑手性非环状α-胺基酮
该反应对各类含有不同取代基的碳酸酯或胺亲核试剂底物具有良好的容忍性。该催化体系可以实现手性α-胺基酮的克级合成,并能保持优秀的对映选择性。生成的胺基酮产物还可以通过不同的官能团转化,得到系列重要的骨架分子。利用该催化体系,可以对商品化的药物分子(如普鲁卡因、苯唑卡因、维生素C)进行衍生化。
该论文第一作者单位及通讯作者单位为西安交大前沿院,前沿院郭武生教授为论文第一作者及唯一通讯作者,前沿院学生左淋洪、崔满营、闫碧维及汕头大学倪绍飞博士参与此项研究。该研究工作得到了西安交通大学启动基金、陕西省和国家高层次人才计划的资助。化合物的表征及测试得到了西安交通大学分析测试共享中心的大力支持。
论文链接:https://doi.org/10.1021/jacs.1c03182
郭武生教授课题组主页:http://gr.xjtu.edu.cn/en/web/wusheng.guo