甲烷作为天然气以及页岩气的主要成分,其高值转化利用是化工领域研究的重要方向。在众多转化路线中,甲烷无氧直接转化为C2烃,即甲烷无氧偶联(如图1所示),因原子经济性高且无二氧化碳排放,具有良好的工业应用前景。目前,虽然二氧化硅晶格限域的单原子铁催化剂,可以在1363 K温度下实现高效甲烷无氧转化,解决了催化剂积碳失活的难题,但是反应温度过高、C2烃选择性较低等问题仍然存在。
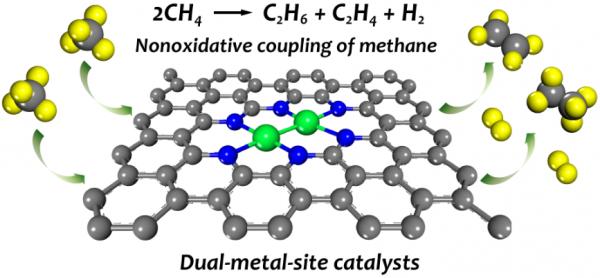
图1 氮掺杂石墨烯限域的双金属位点催化剂用于甲烷无氧偶联反应
近日,西安交通大学化学工程与技术学院常春然教授课题组以提高C–H键活化能力、强化表面C–C键偶联为突破口,采用第一性原理计算、分子动力学模拟、微观动力学模拟等工具,在氮掺杂石墨烯上理论设计出甲烷分解活性高、C2烃选择性高、高温稳定性、抗积碳性能优异的双金属位点催化剂。研究选择非贵金属Fe、Co作为活性中心金属元素,形成两种同核(Fe2–N–C与Co2–N–C)、一种异核(FeCo–N–C)的活性位点。如图2a计算结果所示,三种双金属位点催化剂的C–H键活化能力显著优于单原子催化剂(Fe–N–C与Co–N–C),并且同核位点的活性优于异核位点。经过电子结构分析发现,同核位点金属原子,特别是Co2–N–C,在费米能级位置3dz2的态密度要明显大于异核FeCo–N–C位点的,而高态密度决定了原子的高反应活性。随后,研究计算了两个甲烷分子在三种双金属位点上的无氧偶联反应。计算结果表明,在双金属位点上乙烷会优先生成,并且Co2–N–C的反应活性最高。为了研究反应条件下反应的活性与选择性,研究在Co2–N–C上进行了微观动力学模拟(图2b)。在1200 K温度下,C2产物中乙烷的选择性最高,并且乙烷生成的TOF可以达到10–2s–1,基本满足工业要求水平。此外,从头算分子动力学模拟与反应自由能计算还表明,Co2–N–C催化剂具有良好的高温稳定性与抗积碳能力。因此,本研究预测此类双金属位点催化剂将在甲烷无氧转化过程中具有良好的应用前景。
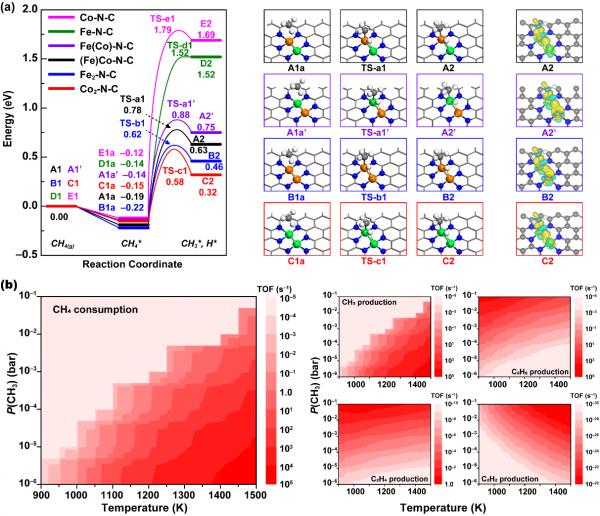
图2甲烷活化与甲烷无氧偶联的理论模拟结果
(a)甲烷在双金属位点与单原子催化剂上的解离势能图以及相应几何结构与电荷密度差分图
(b)甲烷消耗速率与主要产物(CH3、C2H6、C2H4与C2H2)生成速率(转化频率,TOF)
该研究成果以“用于甲烷无氧偶联双金属位点催化剂的理论研究”(Theoretical Insights into Dual-Metal-Site Catalysts for the Nonoxidative Coupling of Methane)为题近日发表在催化领域权威期刊《ACS催化》(ACS Catalysis)。西安交通大学化工学院助理教授黄正清博士为论文第一作者,常春然教授为唯一通讯作者。该项工作得到了国家自然科学基金、中国博士后科学基金、西安交通大学高性能计算平台的支持。
论文链接:https://pubs.acs.org/doi/full/10.1021/acscatal.1c02597
常春然课题组长期从事甲烷无氧转化方面的理论研究,近年来在甲烷无氧转化活性中心的形成机制、动态催化反应机理,以及新型催化剂设计等方面取得了一系列创新性研究成果,为甲烷无氧转化向工业化发展奠定了坚实基础。
论文详情可见常春然课题组主页:http://gr.xjtu.edu.cn/web/changcr









